DeNovoGear: Methods to Identify De Novo Mutations
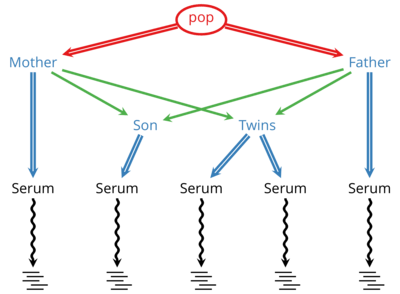
Links: SourceForge — GitHubDeNovoGear (DNG) is developed in a collaboration with the Conrad Lab at Washington University in St. Louis. DNG is an actively developed suite of methods to detect de novo mutations and population variation from sequencing of related individuals and cells.
Using DNG a scientist or clinician can sequence multiple generations in a family and identify locations where germline mutations have occurred in that family. When applied to matched tumor-normal samples, it can identity possible cancer-related somatic mutations.
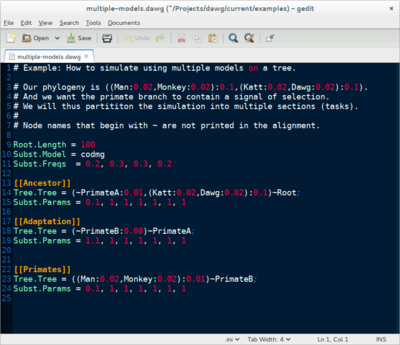
Dawg contains advanced algorithms to simulate the phylogenetic evolution of DNA and an amino acid sequences from a common ancestor. The key innovation of Dawg is biologically accurate models of insertion and deletion.
Dawg 2.0 (beta) offers many new and exciting features. It supports DNA, AA, and codon substitution models. It also supports heterogenous models of evolution, both within a tree and along a sequence. The simulation engine has been completely rewritten and is more efficient than other software.
The beta version is recommended for scientific use. You can download
it from the subversion repository via
svn co svn://scit.us/dawg/current dawg.
Since it is still lacking in documentation, feel free to
ask for help.
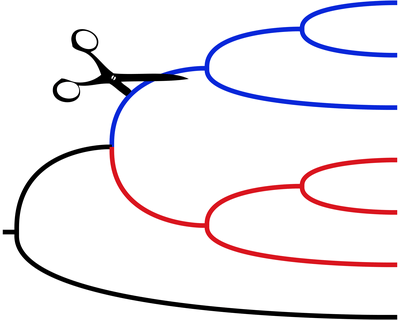
SISRS: SNP Identification from Short Read Sequences
Links: GitHubSISRS — pronounced “scissors” — is a program for identifying phylogenetically informative sites from next-generation whole-genome sequencing of multiple species. It identifies homologous sites without the need to do de novo assembly, annotation, and alignment. It identifies conserved regions by doing joint de novo assembly on multiple species. Sequencing reads are then aligned back to the contigs to identify variable sites.
Ngila uses a statistical model of sequence evolution to find to optimal global alignment between pairs of sequences. It supports biologically realistic power-law models of sequence evolution. In order to find optimal alignments, Ngila implements the Miller and Myers’ (1988) dynamic programming algorithm as well as Hirschberg’s (1975) divide-and-conquer algorithm.

SPAGeDi is a computer package primarily designed to characterize the spatial genetic structure of mapped individuals and/or mapped populations using genotype data of any ploidy level. It was developed by Olivier Hardy and Xavier Vekemans. I maintain a portable/unix source code repository on GitHub.
RedLynx: Population Genetics Simulator
Links: WebsiteRedLynx is a population genetics simulator built entirely in JavaScript. It runs entirely withing a web browser and is extremely helpful when teaching population genetics and evolution. It can simulate genetic drift, selection, dominance, mutation, and migration.
SoFoS: Site Frequency Spectrum Rescaler
Links: WebsiteSoFoS is a Web 2.0 application for rescaling genetic polymorphism data to match a common sample size.
Jak: Coalescent Simulator
Links: GitHubJak is a program that simulates finite-site mutation on coalescent lineages. It focuses on speed of execution.
Klineage: Forward Simulator of Ancestral Lineages
Links: Paper
Klineage is a program that simulates a Wright-Fisher population with
multiplicative fitnesses and finite-site mutation.
It tracks parent-offspring relationships and follows the ancestral
lineage of the population, which is the historical series of
fixed sequences in the population. Klineage can be acceded from
a subversion repository:
svn co svn://scit.us/klineage/current/ klineage.