The Cartwright Lab
The Cartwright Lab is based in the Human and Comparative Genomics Laboratory in the Biodesign Institute at Arizona State University (Tempe, Arizona USA). Our research focuses on various topics in the field of computational evolutionary genetics. We develop methods and software to analyze large genomic datasets and “big data”.
The majority of our research is related to the detection and analysis of mutations and variation from next-generation sequencing. We are species-neutral and work on taxa across the tree of life. Recent work involves humans, cancer, bonobos, ciliates, maize, Plasmodium, Leishmania, E. coli, archaea, Solanaceae, strawberries, and Anolis.
Links: Lab Members — Research Papers — Our Software
A Conspiracy of Minions
Members of the Cartwright Lab come from diverse academic backgrounds. We are biologists, statisticians, mathematicians, engineers, and computer scientists. We are professors, research scientists, postdocs, graduate students, and undergraduate students.
If you are interested in joining the lab, contact me.
The capacity to blunder slightly is the real marvel of DNA. Without this special attribute, we would still be anaerobic bacteria, and there would be no music.
Many a research paper, textbook chapter, and grant proposal has begun with the phrase “Mutation is the ultimate source of genetic variation.” Implicit in this phrase is the assumption that genetic variation is required for evolution. Without mutation, evolution would not be possible, and life itself could never have arisen in the first place.
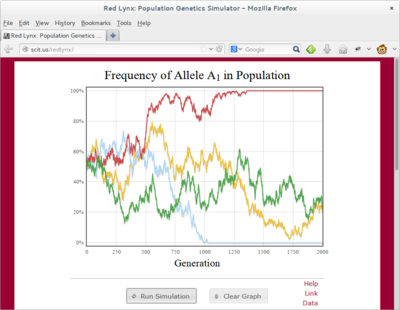
Mutation Accumulation in Tetrahymena thermophila
The ciliate Tetrahymena thermophila is a single-celled protozoan with two nuclei. One nucleus, the macronucleus is transcriptionally active and manages the daily activities of the organism. The other nucleus, the micronucleus, is inactive but is used to produce daughter cells and regenerate macronuclei during sexual reproduction.
In a NIH-funded collaboration with the Zufall and Azevedo labs at the University of Houston, we are studying patterns and phenotypic effects of mutations. Tetrahymena is a nearly perfect system for this research since we can force it to divide asexually which means that mutations that arise on the micronucleus are not expressed or subject to selection.
DeNovoGear: Methods to Identify De Novo Mutations
DeNovoGear (DNG) is developed in a collaboration with the Conrad Lab at Washington University in St. Louis. DNG is an actively developed suite of methods to detect de novo mutations and population variation from sequencing of related individuals and cells.
Using DNG a scientist or clinician can sequence multiple generations in a family and identify locations where germline mutations have occurred in that family. When applied to matched tumor-normal samples, it can identity possible cancer-related somatic mutations.
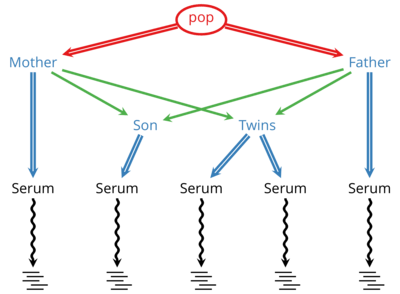
SISRS: SNP Identification from Short Read Sequences
Knowledge of evolutionary relationships form the foundation of biological research. We are developing phylogenetic methods to take advantage of large, whole-genome sequencing to estimate relationships. As sequencing costs decrease, many scientists will need tools that can handle such “big data“.
SISRS, developed by Dr. Rachel Schwartz, is able to identify phylogenetically informative sites from whole-genome datasets. Our technology is extremely useful for people studying emerging infectious diseases and understudied species that lack reference genomes.
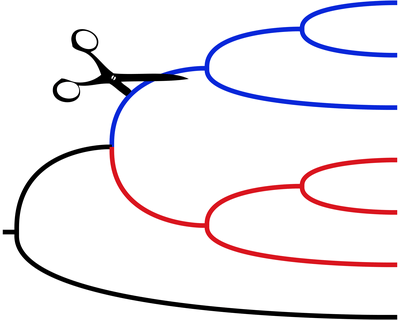
Indel Patterns Across the Tree of Life
Insertions and deletions (indels) are a major class of mutation and play a significant role in molecular evolution. Despite their ubiquity indels are often overlooked in studies because they can be difficult to analyze bioinformatically.
We are interested in developing novel models and approaches to study indels. Our long-term goal is to study indels across the tree of life and determine where the patterns of indel mutation have been conserved and where they have changed.
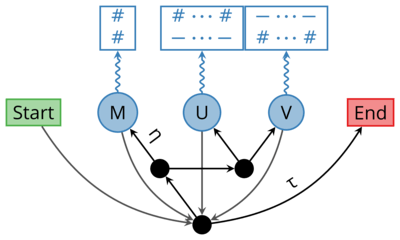
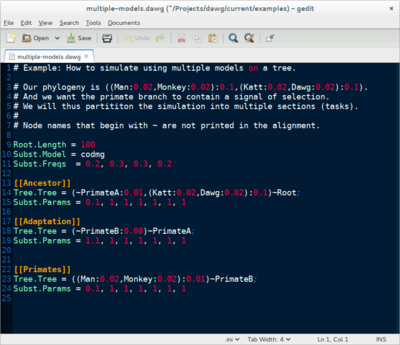
Dawg: Simulating Sequence Evolution
Data simulation is an important tool to test the accuracy of bioinformatic software and validate hypotheses. Dawg contains advanced algorithms to simulate the phylogenetic evolution of DNA and an amino acid sequences from a common ancestor. The key innovation of Dawg is biologically accurate models of insertion and deletion.
Dawg 2.0 (beta) offers many new and exciting features. It supports DNA, AA, and codon substitution models. It also supports heterogenous models of evolution, both within a tree and along a sequence. The simulation engine has been completely rewritten and is more efficient than other software.
Scientific Programming Using JavaScript
JavaScript is the programming language found in all modern web browsers and the workhorse behind Web 2.0. It is a really powerful language for publishing scientific software: GUI design is trivial and it execution occurs on the client’s machine.
We use modern JS libraries like jQuery, jQuery UI, and flot to build interactive scientific and teaching software that lives on the web.